


1 na 7500 żywych urodzeń – taka jest częstość występowania zespołu Williamsa-Beurena. To rzadkie zaburzenie genetyczne wywiera wpływ na wiele układów organizmu, powodując charakterystyczne zmiany w wyglądzie, funkcjonowaniu serca oraz rozwoju poznawczym.
95% osób z tym zespołem ma submikroskopową delecję DNA w regionie długiego ramienia chromosomu 7 (7q11.23). W usuniętym fragmencie chromosomu znajduje się 26–28 genów, wśród których kluczową rolę odgrywa gen elastyny. Charakterystyczny wygląd twarzy, określany jako twarz "elfa", to jeden z najłatwiej rozpoznawalnych objawów tego zespołu.
75-80% pacjentów rozwija wady serca i naczyń krwionośnych. U połowy osób z zespołem występuje nadciśnienie tętnicze, podczas gdy ich profile psychologiczne i poznawcze prezentują fascynujące cechy. Szczególnie uderzającą właściwością jest częste występowanie słuchu absolutnego – zdolności niezwykle rzadkiej w populacji ogólnej.
Zespół Williamsa-Beurena wykracza daleko poza fizyczne objawy. Osoby dotknięte tym zaburzeniem wykazują unikalną kombinację wyzwań rozwojowych i niezwykłych zdolności, które wymagają specjalistycznego podejścia zarówno diagnostycznego, jak i terapeutycznego. Artykuł przedstawia kluczowe aspekty kliniczne, metody rozpoznawania oraz charakterystyczne funkcjonowanie psychospołeczne osób z zespołem Williamsa-Beurena.
Genetyczne podłoże zespołu Williamsa-Beurena
Specyficzna zmiana chromosomowa w chromosomie 7 stanowi podstawę genetyczną zespołu Williamsa-Beurena, prowadząc do szeregu zaburzeń rozwojowych o złożonym charakterze.
Delecja 7q11.23 i utrata genu ELN
Mikrodelecja fragmentu materiału genetycznego na długim ramieniu chromosomu 7, w regionie 7q11.23, jest podstawową przyczyną zespołu Williamsa-Beurena. U 95% osób z tym zespołem wykrywa się hemizygotyczną submikroskopową delecję DNA w tym obszarze. Ubytek obejmuje od 1,5 do 1,8 miliona par zasad, skutkując utratą 26-28 genów.
Gen ELN odgrywa kluczową rolę wśród usuniętych genów. Koduje elastynę – białko budulcowe włókien sprężystych obecnych w wielu tkankach organizmu. Elastyna występuje w płucach, skórze, naczyniach krwionośnych oraz strunach głosowych. Jej niedobór bezpośrednio wyjaśnia charakterystyczne objawy zespołu: nadzastawkowe zwężenie tętnicy głównej, przedwczesne zmarszczki, zachrypnięty głos czy skłonność do przepuklin.
Rola genów GTF2I, LIMK1 i RFC2 w rozwoju mózgu
W regionie objętym delecją znajdują się również inne geny istotne dla prawidłowego rozwoju i funkcjonowania mózgu:
- LIMK1 – koduje kinazę LIM-1 ulegającą ekspresji w mózgu; jej brak prawdopodobnie odpowiada za zaburzone widzenie przestrzenne u pacjentów z zespołem Williamsa
- RFC2 – odpowiedzialny przede wszystkim za niedobór wzrostu oraz problemy rozwojowe
- GTF2I i GTF2IRD1 – geny wpływające na rozwój i funkcje poznawcze
- CLIP2 (dawniej CYLN2) – oddziałuje na funkcjonowanie mózgu
Delecja wszystkich wymienionych genów negatywnie wpływa na rozwój i funkcjonowanie układu nerwowego. Badania wskazują, że utrata genów GTF2I, GTF2IRD1 oraz LIMK1 może odpowiadać za zaburzenia neurobehawioralne charakterystyczne dla osób z zespołem Williamsa-Beurena.
Dziedziczenie i przypadki sporadyczne
Zespół Williamsa-Beurena występuje równie często u obu płci i obserwuje się go na całym świecie, niezależnie od grupy etnicznej. Ubytki genetyczne powstają najczęściej samorzutnie wskutek nierównej rekombinacji chromatyd chromosomu 7 podczas podziału mejotycznego.
Zdecydowana większość przypadków ma charakter sporadyczny – w danej rodzinie nie odnotowuje się innych osób z tym schorzeniem. Te mutacje de novo powstają samoistnie na wczesnym etapie rozwoju prenatalnego.
60% przypadków mikrodelecji ma pochodzenie matczyne, podczas gdy 40% pochodzi od ojca. U niektórych pacjentów delecja może być dziedziczona autosomalnie dominująco. Gdy jeden z rodziców ma mikrodelecję 7q11.23, ryzyko wystąpienia zespołu u każdego dziecka wynosi 50%. Identyczne ryzyko dotyczy potomstwa osoby z zespołem Williamsa-Beurena.
W przypadku więcej niż jednego przypadku zespołu w rodzinie zaleca się badania w kierunku mozaikowatości u rodziców. Gdy choroba występuje jako mutacja de novo, ryzyko jej pojawienia się u kolejnego dziecka tej samej pary rodziców jest niskie (<1%), choć nieco wyższe od ryzyka populacyjnego.
Charakterystyczne objawy kliniczne
Rozpoznanie zespołu Williamsa-Beurena opiera się na specyficznych cechach klinicznych, które ujawniają się już w pierwszych latach życia dziecka. Kombinacja charakterystycznych objawów pozwala lekarzom na wstępną identyfikację tego rzadkiego schorzenia genetycznego.
Zespół Williamsa wygląd twarzy i cechy dysmorficzne
100% pacjentów wykazuje charakterystyczne cechy dysmorficzne twarzy, które stają się wyraźnie widoczne między 1. a 2. rokiem życia. Ze względu na unikalny wygląd, osoby te określane są mianem "dzieci-elfów".
Typowe cechy obejmują:
- wydatne, szerokie czoło z krótkimi brwiami
- duże, szeroko rozstawione oczy z niebieskimi lub zielonymi tęczówkami o "koronkowym" wzorze
- zmarszczka nakątna przypominająca "azjatyckie oczy"
- mały, zadarty nos z zapadniętą nasadą i szerokim czubkiem
- pełne, obwisłe policzki i szerokie usta z grubymi wargami
- mała żuchwa i duża szczęka, nieprawidłowo wykształcone zęby z defektami szkliwa
Ten charakterystyczny zespół cech tworzy rozpoznawalny obraz "twarzy elfa".
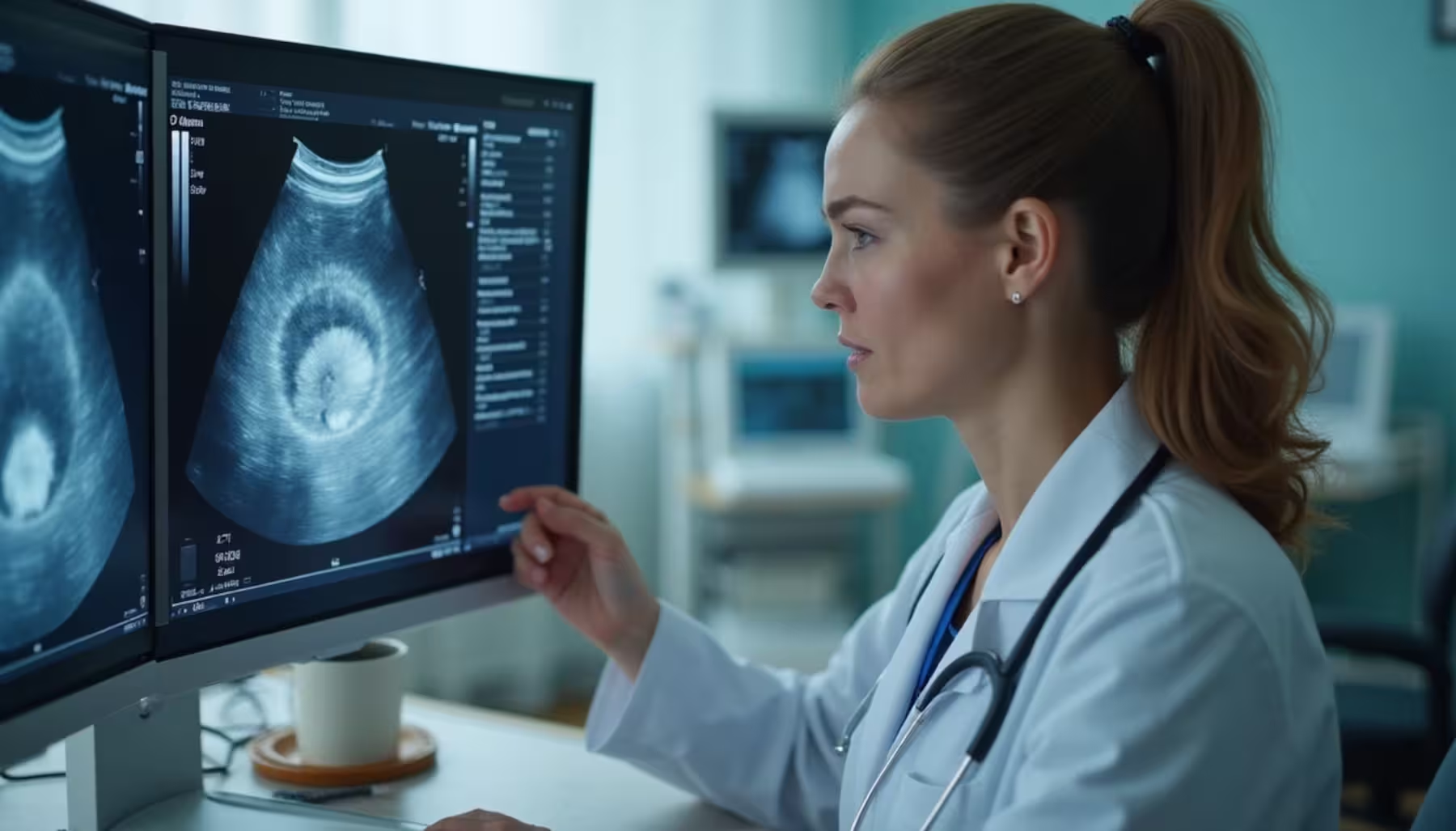
Wady serca: nadzastawkowe zwężenie aorty
Problemy sercowo-naczyniowe dotykają 75-80% osób z zespołem Williamsa-Beurena. Nadzastawkowe zwężenie aorty (SVAS) stanowi najczęstszą wadę, występującą u 75% pacjentów. U niemowląt często obserwuje się również zwężenie obwodowych tętnic płucnych.
Zwężenia mogą objąć różne naczynia – tętnice nerkowe, wieńcowe czy mózgowe. Wada niesie ze sobą wysokie ryzyko nagłego zgonu. 40-50% pacjentów rozwija nadciśnienie tętnicze, które może pojawić się już w dzieciństwie ze względu na zmiany w naczyniach krwionośnych.
Hiperkalcemia i zaburzenia endokrynologiczne
15-30% dzieci z zespołem Williamsa-Beurena doświadcza hiperkalcemii (podwyższony poziom wapnia we krwi) i/lub hiperkalciurii (zwiększone wydalanie wapnia z moczem). Hiperkalcemia najczęściej występuje w okresie noworodkowym i może utrzymywać się do około 5. roku życia. Wielu pacjentów po epizodzie hiperkalcemicznym cierpi z powodu nawracających epizodów.
Osoby z zespołem Williamsa wykazują również podwyższone ryzyko innych zaburzeń endokrynologicznych: nietolerancji glukozy, cukrzycy oraz subklinicznej niedoczynności tarczycy.
Problemy okulistyczne i słuchowe
Około 50% pacjentów rozwija zaburzenia wzroku. Najczęstsze problemy okulistyczne to nadwzroczność i zez zbieżny (27-78% przypadków), podczas gdy u dorosłych przeważa zaćma. Dodatkowe problemy obejmują niedowidzenie (24%) i upośledzone widzenie obuoczne.
Zakażenia ucha środkowego występują często i mogą prowadzić do częściowej utraty słuchu. Charakterystyczna nadwrażliwość na dźwięki może z czasem przekształcić się w głuchotę.
Zespół Williamsa uszy i inne cechy anatomiczne
Uszy mają charakterystyczny wygląd – są nisko osadzone, często powiększone i odstające. Pacjenci wykazują zazwyczaj niski wzrost i wolniejszy rozwój w porównaniu ze zdrowymi rówieśnikami.
Układ moczowo-płciowy może prezentować różne wady: uchyłki pęcherza moczowego, wodonercze, zwężenie tętnicy nerkowej, torbiele nerkowe, a nawet agenezję (brak wykształcenia nerek) lub hipoplazję (niedorozwój nerek).
Przewód pokarmowy również sprawia trudności – niemowlęta mają problemy z przełykaniem, dzieci cierpią na zaparcia i bóle brzucha, a ryzyko celiakii jest podwyższone. Nieprawidłowości tkanek łącznych objawiają się wiotkością więzadeł i nieprawidłową ruchomością stawów.
Diagnoza i metody potwierdzania choroby
Rozpoznanie zespołu Williamsa-Beurena rozpoczyna się od obserwacji charakterystycznych cech fenotypowych oraz wad rozwojowych. Ostateczna diagnoza wymaga jednak specjalistycznych badań genetycznych.
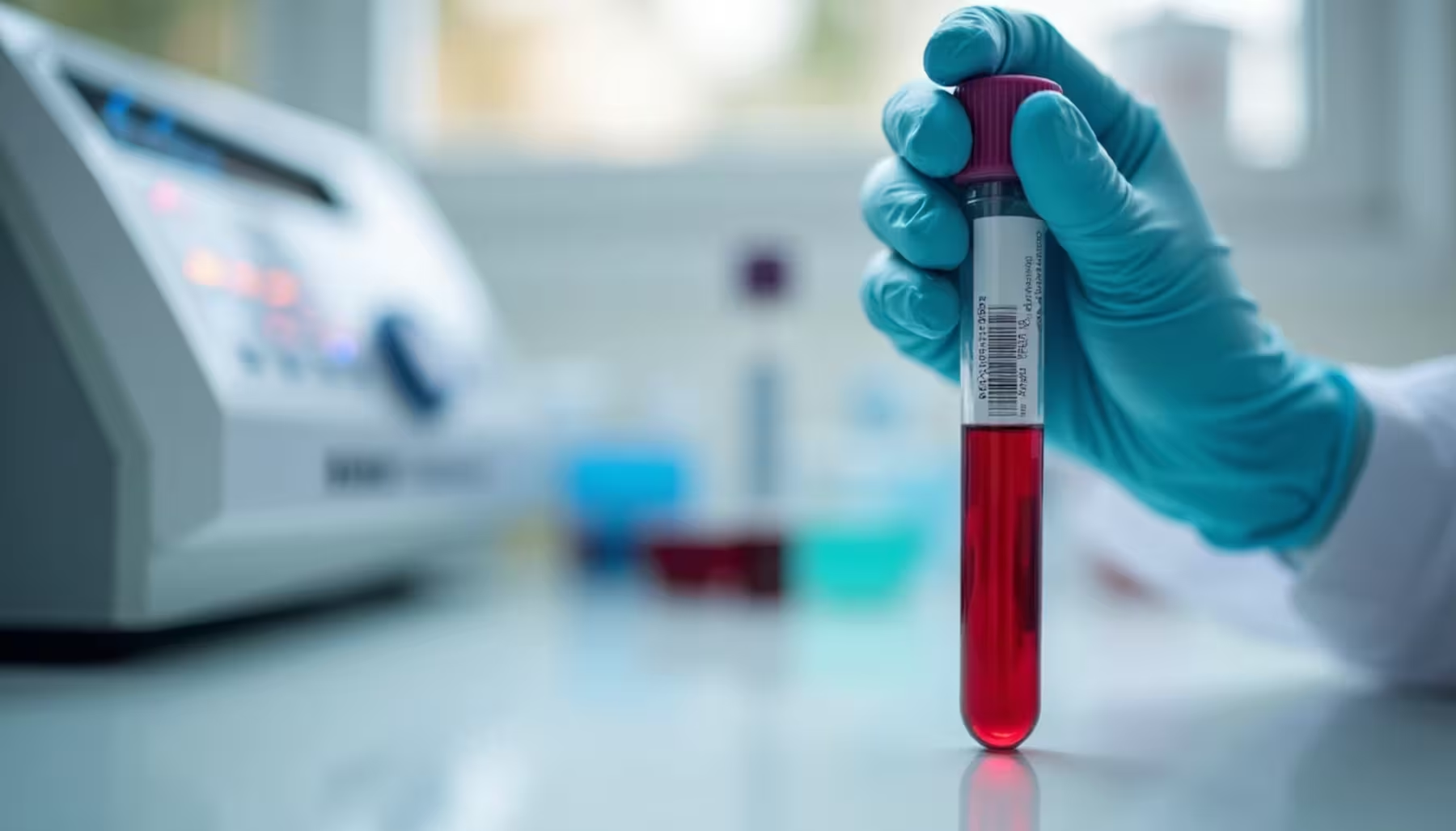
Badanie FISH i wykrywanie mikrodelecji
Technika FISH (fluorescence in situ hybridization) stanowi podstawową metodę potwierdzającą zespół Williamsa-Beurena. Badanie wykorzystuje specjalną sondę, która świeci po przyłączeniu się do poszukiwanego fragmentu chromosomu 7. Brak świecenia wskazuje na delecję tego fragmentu.
99% pacjentów z zespołem Williamsa wykazuje hemizygotyczną submikroskopową delecję chromosomu 7q11.23 wykrywaną techniką FISH. Delecja charakterystyczna dla tego zespołu jest zbyt mała, by można ją było zobaczyć w mikroskopie świetlnym podczas standardowego badania kariotypu.
Dzieci z klinicznymi cechami zespołu Williamsa i negatywnym wynikiem testu FISH wymagają dalszej diagnostyki w poradni genetycznej. Mniej niż 1% osób wymaga uzupełnienia badania o zaawansowane techniki molekularne, takie jak badanie mikromacierzy CGH (arrayCGH) czy badanie MLPA z zastosowaniem sond obejmujących eksony kluczowych genów - ELN, LIMK1 i GTF2I.
Badanie przesiewowe NIFTY Pro
Rodzice planujący potomstwo, szczególnie z wywiadem rodzinnym zespołu Williamsa-Beurena, mogą skorzystać z nieinwazyjnych badań prenatalnych. Test NIFTY Pro należy do grupy testów NIPT (Non-invasive Prenatal Testing).
Od 10 tygodnia ciąży można wykonać test NIFTY Pro poprzez pobranie 10 ml krwi matki. Badanie wykorzystuje sekwencjonowanie nowej generacji (NGS) do analizy pozakomórkowego DNA płodowego (cffDNA) krążącego we krwi ciężarnej. Test identyfikuje różne mikrodelecje i mikroduplikacje.
Ponad 99% wykrywalności przy odsetku wyników fałszywie pozytywnych mniejszym niż 0,05% – to parametry charakteryzujące badanie NIFTY Pro. Szczególnie zalecane jest dla kobiet powyżej 35. roku życia oraz w przypadku występowania chorób genetycznych w rodzinie.
Rola poradni genetycznej i konsultacji rodzinnych
Pacjenci z podejrzeniem zespołu Williamsa-Beurena powinni być kierowani do poradni genetycznej. Doświadczony genetyk kliniczny nie tylko potwierdza rozpoznanie, ale również oszacowuje ryzyko wystąpienia choroby u innych członków rodziny.
Genetyk doradza w kwestii przyszłego planowania rodziny, ocenia przebieg naturalnej historii choroby oraz indywidualizuje podejście terapeutyczne. 50% ryzyko wystąpienia zespołu u każdego dziecka dotyczy przypadków, gdy jeden z rodziców ma mikrodelecję 7q11.23. Większość przypadków występuje jednak sporadycznie jako mutacje de novo.
Diagnostyka różnicowa z innymi zespołami
Zespół Williamsa-Beurena wymaga różnicowania z innymi jednostkami chorobowymi ze względu na podobieństwo niektórych objawów klinicznych. Kluczowe zespoły w diagnostyce różnicowej to:
- zespół Noonana
- zespół Turnera
- zespół delecji 22q11 (zespół DiGeorge'a)
- zespół Smith-Magenis
- zespół Kabuki
- alkoholowy zespół płodowy (FAS)
- zespół Coffina-Lowry'ego
- zespół kruchego chromosomu X
Właściwa diagnoza różnicowa ma fundamentalne znaczenie dla określenia odpowiedniego postępowania terapeutycznego oraz dalszego monitorowania pacjenta. Nowoczesne metody cytogenetyki i biologii molekularnej umożliwiają wyjaśnienie etiologii zaburzeń fenotypowych, nawet gdy standardowe badania nie wykazały nieprawidłowości.
Funkcjonowanie psychospołeczne i poznawcze
75% pacjentów ma zdiagnozowane opóźnienie umysłowe, jednak ich profil psychospołeczny i poznawczy pozostaje wyjątkowy wśród osób z niepełnosprawnością intelektualną. To właśnie specyficzne cechy osobowościowe wyróżniają osoby z zespołem Williamsa-Beurena w sposób niezwykły.
Empatia i nadmierna ufność wobec obcych
95% dzieci określanych jest przez rodziców jako niezwykle przyjacielskie, podczas gdy 98% z łatwością inicjuje kontakty z innymi ludźmi. 88% angażuje się w rozmowy i rozwija je naturalnie, a 90% dzieci nie odczuwa strachu przed obcymi.
Ich zdolność rozpoznawania emocji osiąga poziom stuprocentowej poprawności w testach ekspresji twarzy. Ta niezwykła empatia niesie jednak ze sobą poważne ryzyko – nadmierna ufność wobec nieznajomych czyni te osoby podatnymi na oszustwa i wykorzystanie.
Zdolności muzyczne i pamięć słuchowa
90% dzieci przejawia intensywne zainteresowanie muzyką. Mimo częstej nadwrażliwości na dźwięki (70-90% przypadków), ich uzdolnienia muzyczne są niezwykłe:
- 87% lubi śpiewać, a 86% łatwo zapamiętuje piosenki
- Słuch absolutny występuje u 30-98% osób z zespołem, podczas gdy w populacji ogólnej dotyczy jedynie 1 osoby na 10 000
Trudności w nauce i koordynacji wzrokowo-przestrzennej
Dobrze rozwinięta pamięć słuchowa i zdolności językowe kontrastują z poważnymi deficytami w funkcjach wzrokowo-przestrzennych. Uszkodzenia prawej półkuli mózgu przy względnie zachowanej lewej półkuli powodują szczególne trudności w rysowaniu, czytaniu, matematyce i koordynacji ruchowej.
Zaburzenia snu i lęki nocne
Problemy ze snem dotyczą znacznej części pacjentów – trudności z zasypianiem, nocne wybudzenia oraz niepokój w trakcie snu. Dzieci wymagają obecności dorosłych podczas zasypiania, wykazują mimowolne ruchy nóg, budzą się z płaczem i krzykiem. 73% dorosłych z zespołem rozwija zaburzenia lękowe.
Zespół Williamsa znane osoby – wpływ medialny
Osoby z zespołem Williamsa wnoszą do społeczeństwa wyjątkową wartość poprzez swoją naturalną serdeczność i otwartość. Brak zrozumienia ich specyficznych potrzeb często prowadzi jednak do nieporozumień społecznych. Ich zachowanie bywa określane jako "koktajlowe" – potrafią oczarować rozmówcę już od pierwszego kontaktu, będąc opisywane jako "zawsze wesołe, posiadające czarujące cechy charakteru i niesamowity entuzjazm".
Opieka, leczenie objawowe i długość życia
Zespół Williamsa-Beurena wymaga wyłącznie leczenia objawowego. Nie istnieje obecnie terapia przyczynowa tego rzadkiego schorzenia genetycznego. Skuteczne postępowanie opiera się na kompleksowym podejściu i stałym monitorowaniu pacjenta.
Wielospecjalistyczna opieka: kardiolog, endokrynolog, logopeda
Diagnoza zespołu oznacza konieczność objęcia pacjenta opieką wielospecjalistyczną. Regularne konsultacje kardiologiczne pozostają priorytetem, nawet gdy wcześniejsze badania nie wykazały nieprawidłowości. Pacjenci z zespołem Williamsa mają zwiększone ryzyko nagłej śmierci sercowej, dlatego każdy zabieg w znieczuleniu ogólnym wymaga wcześniejszej konsultacji specjalistycznej.
Opieka endokrynologiczna koncentruje się na monitorowaniu poziomu wapnia i funkcji tarczycy. Równie istotne są regularne wizyty stomatologiczne, neurologiczne i nefrologiczne. Logopeda odgrywa kluczową rolę w usprawnianiu artykulacji i rozwoju komunikacji.
Rehabilitacja metodą NDT-Bobath i SI
Metoda NDT-Bobath pokazuje szczególną skuteczność w terapii dzieci z zespołem Williamsa-Beurena. Można ją naturalnie włączyć w codzienny rytm funkcjonowania. Rehabilitacja skupia się na regulacji napięcia mięśniowego i ułatwieniu prawidłowych wzorców ruchowych.
Równowaga i koordynacja wymagają szczególnej uwagi, ponieważ są zazwyczaj znacznie upośledzone. Zajęcia przy muzyce wykorzystują naturalną muzykalność pacjentów. Integracja sensoryczna stymuluje rozwój umiejętności wzrokowo-przestrzennych, stanowiąc istotny element terapii.
Zespół williamsa długość życia
Długość życia osób z zespołem Williamsa różni się w zależności od źródła danych. Niektóre badania wskazują na skrócenie życia o 10-20 lat w porównaniu z populacją ogólną. Inne źródła podają średnią długość życia na poziomie 50-55 lat, z możliwością dożycia 70 lat.
Jakość opieki medycznej stanowi kluczowy czynnik wpływający na długość życia. Wczesne wykrywanie i leczenie powikłań sercowo-naczyniowych może znacząco poprawić rokowanie.
Rola rodziny i wsparcia społecznego
Rodzina stanowi fundament skutecznej terapii osoby z zespołem Williamsa. Od 2007 roku w Polsce działa Stowarzyszenie Pomocy Osobom z zespołem Williamsa, oferujące wsparcie, pomoc diagnostyczną i rehabilitacyjną. Organizacja umożliwia wymianę doświadczeń między rodzinami dotkniętymi tym schorzeniem.
Wsparcie emocjonalne ze strony bliskich i społeczności lokalnej znacząco wpływa na jakość życia pacjentów. Edukacja rodziców w zakresie prawidłowej pielęgnacji i stymulacji rozwoju dziecka pozostaje istotnym elementem kompleksowej terapii.
Wnioski
Zespół Williamsa-Beurena przedstawia złożony obraz genetycznego zaburzenia, które łączy w sobie wyzwania medyczne z fascynującymi zdolnościami poznawczymi. Delecja regionu 7q11.23 tworzy unikalny wzór objawów – charakterystyczne rysy twarzy współistnieją z problemami sercowo-naczyniowymi, podczas gdy ograniczenia intelektualne kontrastują z wyjątkowymi zdolnościami muzycznymi i społecznymi.
Wczesne rozpoznanie zmienia perspektywy terapeutyczne. Technika FISH pozostaje podstawowym narzędziem diagnostycznym, choć nowoczesne metody molekularne rozszerzają możliwości identyfikacji zespołu. Każdy przypadek wymaga zindywidualizowanego podejścia ze względu na różnorodność prezentacji klinicznej.
Osoby z zespołem Williamsa łączą niepełnosprawność intelektualną z niezwykłą empatią i uzdolnieniami muzycznymi. Ta dychotomia – między ograniczeniami a talentami – definiuje ich codzienne funkcjonowanie. Nadmierna ufność wobec obcych stanowi jednocześnie ich siłę społeczną i potencjalne zagrożenie, wymagając stałego wsparcia edukacyjnego.
Kompleksowa opieka medyczna obejmuje monitoring kardiologiczny, endokrynologiczny oraz neurologiczny przez całe życie pacjenta. Regularne kontrole umożliwiają wczesne wykrywanie powikłań i znacząco wpływają na jakość oraz długość życia.
Społeczna świadomość zespołu pozostaje ograniczona. Organizacje jak Stowarzyszenie Pomocy Osobom z zespołem Williamsa pełnią kluczową rolę w edukacji społecznej i wsparciu rodzin. Połączenie profesjonalnej opieki medycznej z zaangażowaniem społecznym i zrozumieniem wyjątkowych cech osobowościowych umożliwia osobom z zespołem Williamsa osiąganie satysfakcjonującej jakości życia.
FAQ: najczęściej zadawane pytania
Jakie są główne objawy zespołu Williamsa-Beurena?
Główne objawy to charakterystyczny wygląd twarzy ("twarz elfa"), wady serca (zwłaszcza zwężenie aorty), problemy ze wzrokiem i słuchem, opóźnienie rozwoju oraz specyficzne cechy osobowości, takie jak nadmierna towarzyskość i uzdolnienia muzyczne.
Jak diagnozuje się zespół Williamsa-Beurena?
Diagnoza opiera się na obserwacji charakterystycznych cech klinicznych, a następnie jest potwierdzana badaniem genetycznym FISH, które wykrywa delecję w chromosomie 7. W niektórych przypadkach stosuje się również bardziej zaawansowane techniki molekularne.
Czy zespół Williamsa-Beurena jest dziedziczny?
Większość przypadków (około 95%) to mutacje spontaniczne. Jednak jeśli rodzic ma zespół Williamsa, ryzyko przekazania go dziecku wynosi 50%. Dlatego ważne są konsultacje genetyczne dla rodzin dotkniętych tym schorzeniem.
Jakie są charakterystyczne cechy psychospołeczne osób z zespołem Williamsa?
Osoby te cechują się niezwykłą towarzyskością, empatią i zamiłowaniem do muzyki. Często mają trudności z nauką i koordynacją wzrokowo-przestrzenną, ale wykazują dobre zdolności językowe i pamięć słuchową. Charakterystyczna jest też nadmierna ufność wobec obcych.
Jak wygląda leczenie i opieka nad osobami z zespołem Williamsa-Beurena?
Leczenie ma charakter objawowy i wymaga wielospecjalistycznej opieki, obejmującej m.in. kardiologa, endokrynologa i logopedę. Kluczowa jest wczesna rehabilitacja, zwłaszcza metodą NDT-Bobath i integracji sensorycznej. Ważne jest również wsparcie rodziny i odpowiednia edukacja dostosowana do potrzeb pacjenta.
Czy osoby z zespołem Williamsa-Beurena mogą prowadzić samodzielne życie?
Samodzielność osób z zespołem Williamsa-Beurena zależy od stopnia nasilenia objawów i wsparcia, jakie otrzymują. Wiele z nich wymaga pomocy w codziennym funkcjonowaniu, jednak przy odpowiedniej terapii, edukacji i wsparciu środowiskowym możliwe jest osiągnięcie pewnego stopnia niezależności, zwłaszcza w dorosłości.
Jakie trudności edukacyjne najczęściej występują u dzieci z zespołem Williamsa-Beurena?
Najczęstsze trudności dotyczą nauki matematyki, czytania ze zrozumieniem oraz zadań wymagających myślenia abstrakcyjnego i koordynacji wzrokowo-przestrzennej. Dzieci te potrzebują indywidualnego podejścia edukacyjnego, a najlepiej sprawdzają się metody oparte na multisensorycznym uczeniu i pozytywnej motywacji.












.avif)
































































































